Quantum Magnetism
|
|
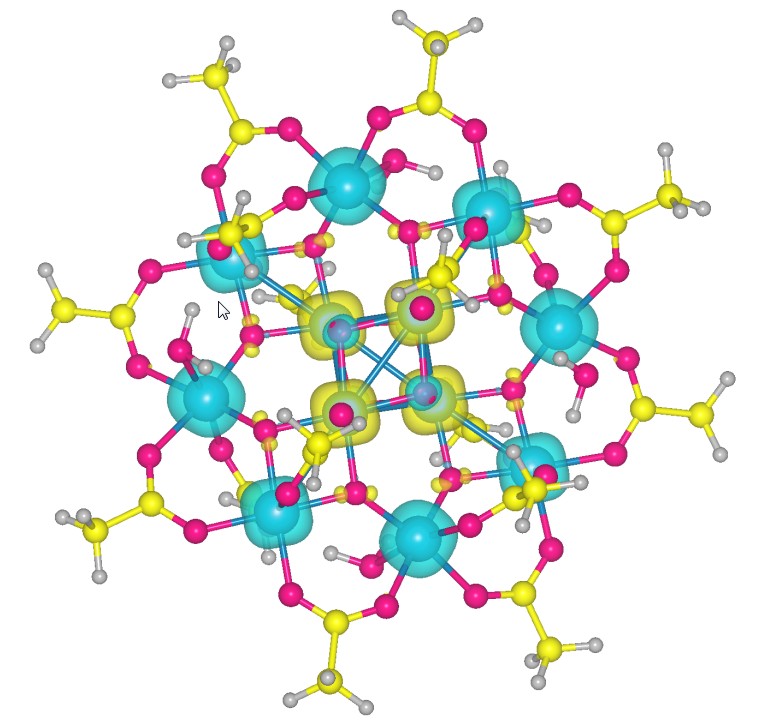
Mn12 molecule with its spin distribution. Majority spin density displays as blue, minority displays as yellow)
|
I am also fascinatied by magnetic properties of the quantum systems at nano-scale. One of the systems I studied is the single molecular magnet Manganese-12-Acetate (Mn 12). The electronic structure of the Mn 12 molecule is reproduced with using DFT+U.
The interaction potential of the molecule with a layer of Boron nitride calculated with DFT indicates that the molecule is physisorbed on the layer. Together with experimentalists, we demonstrated that it is possible to deposite Mn 12 molecules on a insulating substrate so that the magnetic properties of thess fraggle molecules preserve [1]. This work is highlighted on Nature Material [2].
[1] S. Kahle, Z. Deng, N. Malinowski,C. Tonnoir, A. FormentAliaga, N. Thontasen, G. Rinke, D. Le, V. Turkowsky, T. S. Rahman, S. Rauschenbach, M. Ternes, and K. Kern, Nano Lett. 12, 518 (2012).
[2] J. Heber, Nature Mater. 11, 95 (2012).
Catalysis
|
|
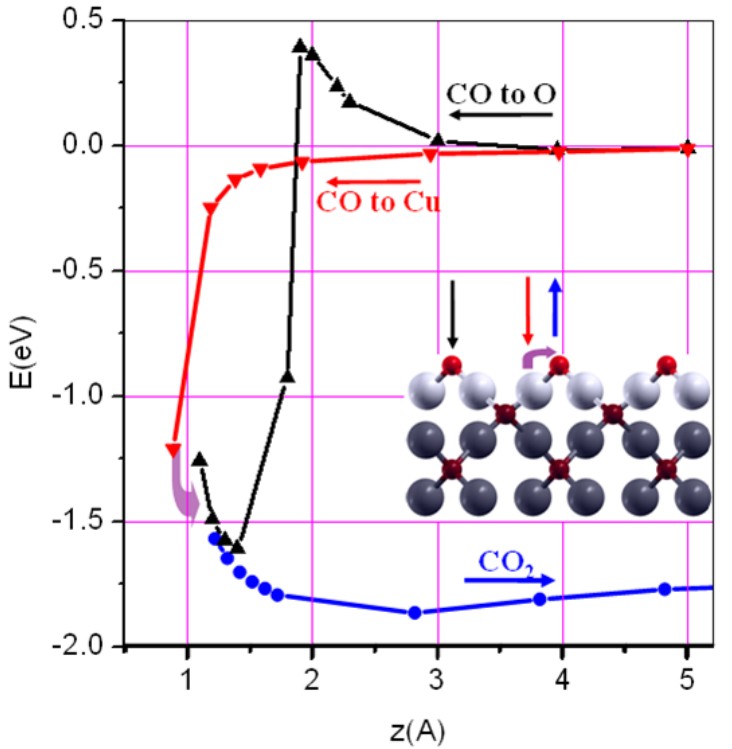
Energertics pathways of CO adsorption and oxidation on Cu2O(100).
|
Catalysis is one of my research interests. I have carried out a systematic first-principles simulations of
the energetic pathways of the CO oxidation on the Cu 2O(100) surface [1, 2]. These simulations
show CO to oxidize spontaneously on the O-terminated Cu 2O(100) surface by consuming
surface oxygen atoms. The O-vacancy on Cu 2O(100) then is subsequently healed by disso-
ciative adsorption of atmospheric O 2 molecules. In collaboration with Dr. Hong, we perfomed KMC simulation for the above reaction [3].
Recently, we are focusing on the design molybdenum sulfide structure for catalitic purposes [4].
[1] B. White, M. Yin, A. Hall, D. Le, S. Stolbov, T. Rahman, N. Turro, and S. O'Brien, Nano Lett. 6, 2095 (2006).
[2] D. Le, S. Stolbov, and T. S. Rahman, Surf. Sci. 603, 1637 (2009).
[3] S. Hong, D. Le, and T. S. Rahman. In preparation.
[4] D. Sun, W. Lu, D. Le, Q. Ma, M. Amanpour, M. Alcántara Ortigoza, S. Bobek, J. Mann, J. Wyrick, T. S. Rahman, L. Bartels, Angew. Chem. Int. Ed. 51, 10284 (2012)
Molybdenum sulfilde on Cu(111)
|
|
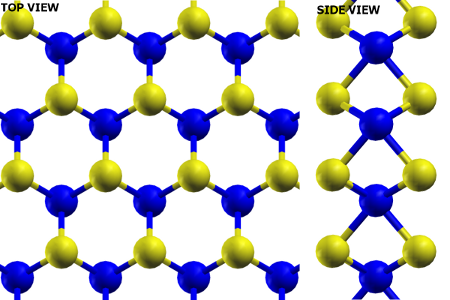
Single layer MoS2
|
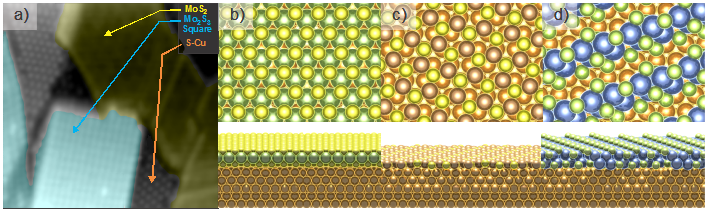
Single layer molybdenum disulfide (MoS 2) attracts our attention because it is a prominent material for future applications, owing to its direct band-gap of ~1.9 eV. One challenge is to be able to produce large-high-quality sheet of MoS 2. Together with Bartels's group, we have devoted significant effort to grow and have succesfully grown reasonably large patches of MoS 2 on Cu(111). Our results show the regular Morié pattern of MoS 2 grown on Cu(111) with periodicity of about 1.3 nm [1].
|
|
Simulated STM image of MoS2/Cu(111)
|
Our first-principles calculations [2] of the geometric and electronic structures of a single layer MoS 2 on Cu(111) utilizing the van der Waals density functional show three energetically equivalent stacking types and a Moiré pattern whose periodicity is in agreement with experimental findings. The layer is found not to be purely physisorbed on the surface, rather there exists a chemical interaction between it and the Cu surface atoms. We also find that the MoS 2 film is not appreciably buckled, while the top Cu layer gets reorganized and vertically disordered.
|
|
STM image of MoSx structures and thier models on Cu(111).
|
Although MoS 2 basal plane is quite inert, MoS 2 nano-particles are good catalysts for hydrodesulfurization and hydrodenitrogenation processes. The high catalytic activity may be ascribed to a metallic electronic state at the brim of MoS 2 triangular clusters. On the path to explore the growth of MoS x, we have recently discovered a new molybdenum sulfide structure, grown on Cu(111). The new structure coexists with MoS 2 patches and triangular islands on Cu(111). More importantly, we have shown the high catalytic activity of the new structure [3].

|
|
STM image and model of Mo6S6 nanowires on Cu(111).
|
Based on DFT predictions and STM measurements we report the possibility of using the Cu(111) surface for growth of molybdenum sulfide nanowires (Mo 6S 6) [4]. Strong substrate interactions coupled with small lattice mismatch lead to epitaxial growth of the nanowires parallel to a set of substrate high symmetry directions. We observe a propensity for creation of aligned and equally spaced arrays of nanowires and use DFT to elucidate interaction strength both in the absence and presence of the substrate.
[1] D. Kim, D. Sun, W. Lu, Z. Cheng, Y. Zhu, D. Le, T. S. Rahman, L. Bartels, Lanmuir 27, 11650 (2011).
[2] D. Le, D. Sun, W. Lu, L. Bartels, T. S. Rahman, Phys. Rev. B 85, 075429 (2012).
[3] D. Sun, W. Lu, D. Le, Q. Ma, M. Aminpour, M. Alcántara Ortigoza, S. Bobek, J. Mann, J. Wyrick, T. S. Rahman, L. Bartels, Angew. Chem. Int. Ed. 51, 10284 (2012)
[4]D. Le, D. Sun, W. Lu, M. Aminpour, C. Wang, Q. Ma, T.S. Rahman, and L. Bartels, Surf. Sci. In press (2013).
van der Waals interactions
In general, density functional theory (DFT), whether with the local density approximation (LDA) or with a generalized gradient approximation (GGA), is not expected to provide an accurate description of the van der Waals (vdW) interactions, owing to the built-in
locality of their exchange-correlation functionals. One of the current challenges is to accurately account for the vdW forces (London dispersion forces) in DFT simulations
. Over the years, many schemes have been proposed for incorporating vdW interactions
into DFT calculations.
The variety of the methods gives rise to questions of how the results obtained from
each method compare and of, more importantly, how the inclusions of vdW interaction effects
conclusions of studies of the systems under consideration.
Through the case of the adsorption of DNA nulceobases on graphene and of n-alkanes on Pt(111), we showed the serious discrepancies in the results obtained by certain widely used methods for
incorporating vdW interactions [1].
We explored the role of vdW interactions in pattern formation of molecules on
catalyst surfaces.
We demonstrated that the vdW is essential for the pattern formation and for elastic property of anthracene film on Cu(111) and that the competition between the different adsorption sites and configurations leads to the formation of various adsorbate structures [2]. 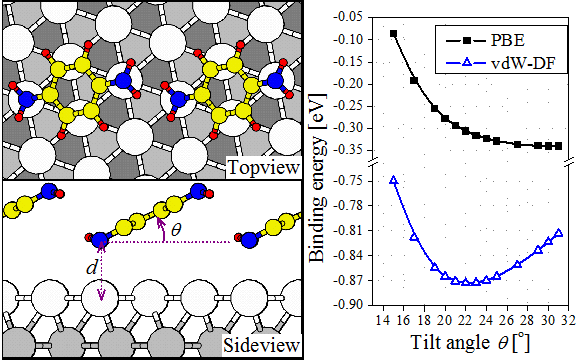 |
| Model of BDA line-structure on Au(111) |
We showed that the inclusion of vdW interactions between 3 amine moleculeis and the Au(111) surface leads to good agreement with experimental data on the binding energies. Each molecule adsorbs with a small tilt angle. For the case of 1,4-diaminobenzene (BDA) our calculations reproduce the larger tilt angle measured by photoemission experiments, when intermolecular interactions are included. These results point to not only the importance of vdW interactions to molecule-surface binding, but also that of intermolecular interactions in determining the adsorption geometry and pattern formation [3].
[1] D. Le, PhD Dissertation (2012).
[2] D. Sun, D. Kim, D. Le et al, Phys. Rev. B 82, 201410(R) (2010).
[3] D. Le, M. Aminpour, A. Kiejna and T. S. Rahman, J. Phys.: Condens. Matter 24, 222001 (2012).
Optical properties of nano-materials
One of my research interest is the optical properties of nano-materials. I am involved in the research of the "Linker-Induced Anomalous Emission of Organic-Molecule Conjugated Metal-Oxide Nanoparticles" [1].
|
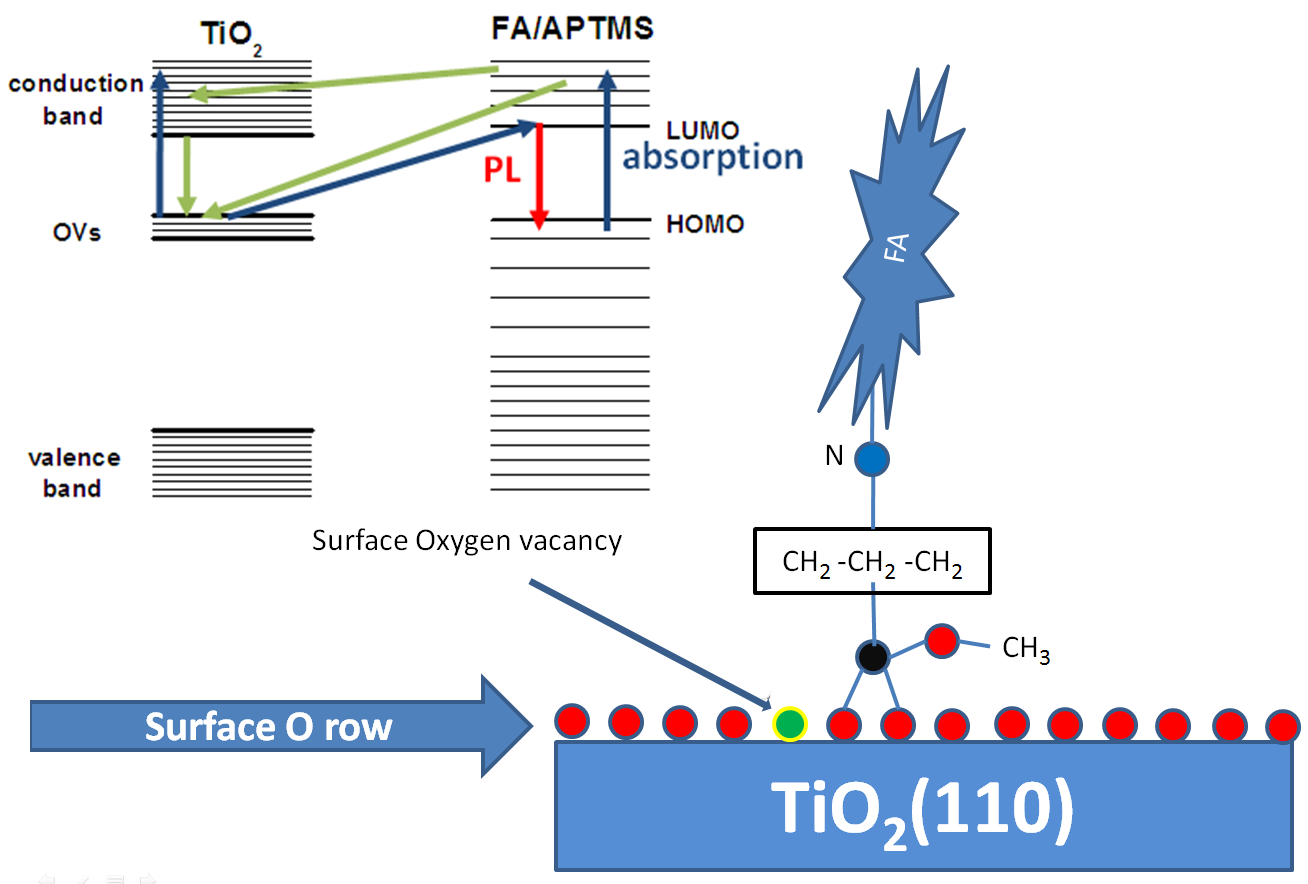
Scheme of PL of FA/APTMS/TiO2 system
|
Semiconductor nanoparticles conjugated with organic- and dye-molecules to yield high efficiency visible photoluminescence (PL) hold great potential for many future technological applications. We show that folic acid (FA)-conjugated to nanosize TiO 2 and CeO 2 particles demonstrates a dramatic increase of photoemission intensity at wavelengths between 500 and 700 nm when derivatized using aminopropyl trimethoxysilane (APTMS) as spacer-linker molecules between the metal oxide and FA. Using density-functional theory (DFT) and time-dependent DFT calculations we demonstrate that the strong increase of the PL can be explained by electronic transitions between the titania surface oxygen vacancy (OV) states and the low-energy excited states of the FA/APTMS molecule anchored onto the surface oxygen bridge sites in close proximity to the OVs. We suggest this scenario to be a universal feature for a wide class of metal oxide nanoparticles, including nanoceria, possessing a similar band gap (3 eV) and with a large surface-vacancy-related density of electronic states. We demonstrate that the molecule-nanoparticle linker can play a crucial role in tuning the electronic and optical properties of nanosystems by bringing optically active parts of the molecule and of the surface close to each other.
[1] V. Turkowski, S. Babu, D. Le, MK. Haldar, A. Wagh, Z. Hu, A. S. Karakoti, A. J. Gesquiere, B. Law, S. Mallik, T. S.Rahman, M. N. Leuenberger, S. Seal, ACS Nano 6, 4854 (2012)..
|