
Talat S. Rahman
UCF Trustee Chair Professor and Pegasus Professor
Department of Physics
University of Central Florida
- Multi-scale modeling of chemical reactions and related phenomena at surfaces
- Understanding processes that control growth and morphological evolution of thin films
- Theory and modeling of vibrational, optical and magnetic properties of nanomaterials
- Predictive modeling of functional two-dimensional transition metal dichalcogenides
- Surface coordination chemistry: novel functionality via substrate charge transfer and oxidation state
- Understanding the response of surfaces and nanostructures to ultrafast external fields
- Development of techniques beyond density functional theory for strongly correlated material
- Development of techniques suitable for non-equilibrium phenomena and non-adiabatic processes
A DFT+DMFT approach for nanosystems
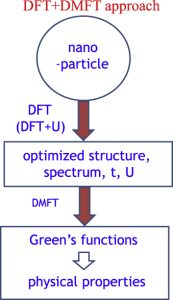
We propose a combined density-functional-theorydynamical-mean-field-theory (DFT + DMFT) approach for reliable inclusion of electronelectron correlation effects in nanosystems. Compared with the widely used DFT + U approach, this method has several advantages, the most important of which is that it takes into account dynamical correlation effects. The formalism is illustrated through different calculations of the magnetic properties of a set of small iron clusters (number of atoms 2 ≤ N ≤ 5). It is shown that the inclusion of dynamical effects leads to a reduction in the cluster magnetization (as compared to results from DFT + U) and that, even for such small clusters, the magnetization values agree well with experimental estimations. These results justify confidence in the ability of the method to accurately describe the magnetic properties of clusters of interest to nanoscience.
This work has been reported as a FAST TRACK COMMUNICATION on the Journal of Physics: Condensed Matter [V. Turkowski, A. Kabir, N. Nayyar and T. S. Rahman, J. Phys.: Condens. Matter 22 462202 (2010)]
A Surface Coordination Network Based on Substrate-Derived Metal Adatoms with Local Charge Excess
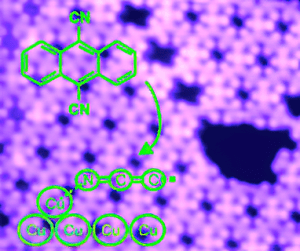
We present a coordination network, formed at a metal surface in a system facile to prepare because the required metal centers can be released in a measured fashion from the substrate by simple annealing. Analysis of the charge density in this system suggests that metal adatoms can have a pronounced donating character despite the electron deficiency of uncoordinated adatoms. The notion that metal adatoms present an anionic character in their interaction with molecular adsorbates could have significant and widespread implications (for example, in heterogeneous …
This work paper has been reported on the Angewandte Chemie (International Edition) [Greg Pawin et al, Angew. Chem. Int. Ed. 47, 8442 (2008)]
Toward an Understanding of Ligand Selectivity in Nanocluster Synthesis
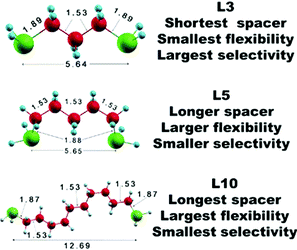
We performed scalar relativistic density functional theory (DFT) calculations using the projector augmented wave scheme (PAW) to examine the reactivity and selectivity of diphosphine ligands LM, with the formula PH2(CH2)MPH2 (spacer M = 3, 5), toward small-sized cationic Aun (n = 7-11) nanoclusters. By isolating the ligand-induced contribution to the stability condition, we show that such interaction selectively stabilizes the cationic Au11 cluster. Furthermore, we find that L5
This work has been reported on the Journal Of Physical Chemistry C [S. Hong, G. Shafai, M. Bertino, and T. S. Rahman, J. Phys. Chem. C 115 14478 (2011)]
Time-dependent density-matrix functional theory for biexcitonic phenomena
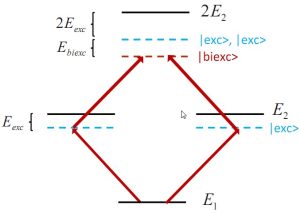
We formulate a time-dependent density-matrix functional theory (TDDMFT) approach for higher-order correlation effects like biexcitons in optical processes in solids based on a reduced two-particle density-matrix formalism within the normal orbital representation. A TDDMFT version of the Schrodinger equation for biexcitons in terms of one- and two-body reduced density matrices is derived, which leads to finite biexcitonic binding energies already with an adiabatic approximation. Biexcitonic binding energies for several bulk semiconductors are…
This work has been reported on the Physical Review B [V. Turkowski, C. A. Ullrich, T. S. Rahman, and M. N. Leuenberger, Phys. Rev. B 82 205208 (2010)]
Selective oxidation of ammonia on RuO2(110)
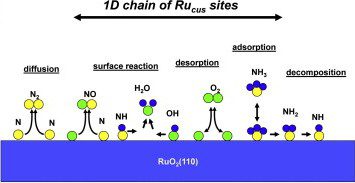
We have used a combination of density functional theory (DFT) and kinetic Monte Carlo (KMC) simulations to calculate the reaction rates for the selective oxidation of ammonia on RuO2(110). Our KMC simulations of 18 reactions among NHx (x=0-3) and OHx (x=0-2) species on RuO2(110) show 93% selectivity for NO, in close agreement with experiment (~95%). The chief factor in the high selectivity for NO on the RuO2(110) surface is the significantly reduced N diffusion (via N blocking) caused …
This work has been reported on the Journal of Catalysis [S. Hong, A. Karim, T. S. Rahman, K. Jacobi, G. Ertl, J. Catal. 276 371 (2010)]
Off-lattice pattern recognition scheme for KMC simulations

We report the development of a pattern-recognition scheme for the off-lattice self-learning kinetic Monte Carlo (KMC) method, one that is simple and flexible enough that it can be applied to all types of surfaces. In this scheme, to uniquely identify the local environment and associated processes involving three-dimensional (3D) motion of an atom or atoms, space around a central atom is divided into 3D rectangular boxes. The dimensions and the number of 3D boxes are determined by the accuracy with which a process needs to be identified and a process is described as the central atom moving to a neighboring vacant box accompanied by the motion of any other atom or atoms in its surrounding boxes.
This work has been reported on the Journal of Computational Physics [G. Nandipati, A. Kara, S. I. Shah, T. S. Rahman, J. Comp. Phys. 231 3548 (2012)]
Role of vdW in the tilted binding of amine molecules to Au(111)
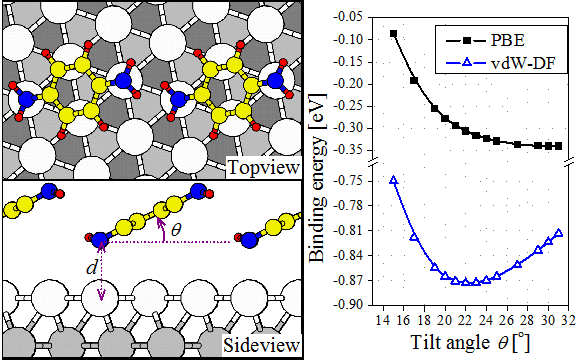
We show that the inclusion of van der Waals interactions between the isolated molecule (3 amine molecules) and the Au(111) surface leads to good agreement with experimental data on the binding energies. Each molecule adsorbs with a small tilt angle. For the case of 1,4-diaminobenzene (BDA) our calculations reproduce the larger tilt angle measured by photoemission experiments, when intermolecular interactions are included. These results point to not only the importance of vdW interactions to molecule-surface binding, but also that of intermolecular interactions in determining the adsorption geometry and pattern formation.
This work has been reported as a FAST TRACK COMMUNICATION on the Journal of Physics: Condensed Matter [D. Le, M. Aminpour, A. Kiejna and T. S. Rahman, J. Phys.: Condens. Matter 24 222001 (2012)]
